
有种贫血病,会致癌
文章来源:向日葵儿童 作者:徐佳琳 责任编辑:hanping 时间:2020-07-13
贫血是种常见的病症,但许多人不知道的是,有一种贫血比较罕见却非常危险,甚至能让孩子得上癌症.......
被“诅咒”的家庭
拿着小儿子的血检报告,王萍的心情难以平复。这已经是家中第三个出现这种病症的孩子了。
孩子们出生后不久,王萍就发现自家几个儿子的头围都比别家孩子小。她有些担心,但孩子们智力发育都挺正常,似乎和其他孩子没啥差别。
但不久,老大就生病了,血检报告显示红细胞体积增大,并出现溶血。医生查了很久,但直至孩子病危时也没查出个所以然,最后说是严重贫血。
没过几年,老二也发生了同样的疾病。这回医生们吸取了经验,在病危前确诊了贫血,却无济于事,回天乏术。
而如今,在死了两个孩子后,家中的小儿子在5岁多也逐渐出现了同样的症状:皮肤上出现大量褐色斑块,常常出血、生殖器发育不全、斗鸡眼……三个孩子接二连三在5-7岁间被怪病缠身而去世。这一家是被命运诅咒了吗?
“怪病”范可尼贫血
影响这家三个孩子的并非什么诅咒,而是一种叫“范可尼贫血”的罕见病。
范可尼贫血是一种家族性遗传病,主要影响骨髓,表现为各种类型血细胞均有减少。它是所有遗传性再生障碍性贫血中最常见的一类,大概每20到40万个人里会有一个人患病。
20世纪20年代末,瑞士儿科专家范可尼医生发现,有一家三兄弟和王萍的三个儿子一样,在出生后都出现了不同程度的发育障碍,并先后死于严重贫血。
范可尼医生很快意识到,患者不仅所有血细胞都会因这种病而减少,还容易早早患上各种癌症,尤其是骨髓增生异常综合征和急性粒细胞白血病。
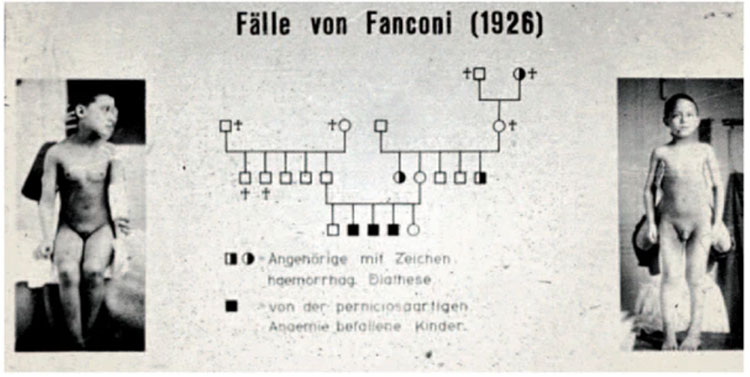
图2 范可尼医生对最初病例和疾病家族遗传谱系的研究报告
根据病人的家族病史,范可尼医生推断出这是一种遗传病,但造成这种遗传病的究竟是什么?没有人知道。
经过大半个世纪的研究,科学家们才发现,这种遗传“怪病”的种种症状是由细胞中大量的染色体畸形引起的,导致这些染色体畸形的根本原因是基因突变——而且不止一个。
迄今为止,科学家们已经找到了22个和范可尼贫血有关的基因,这些基因的突变都可能引起范可尼贫血。
这22个基因都和遗传物质的修复有关,其中一个叫做BRCA2(也叫FANCD1),是一个和多种癌症有关的基因,也是这22个基因中最广为人知的之一。
正常人体内的基因都有两份,一份来自父亲,一份来自母亲。以BRCA2基因为例,如果孩子的两份 BRCA2基因都有突变,那么遗传物质出错后,就没法像正常人那样得到修复了。
于是,孩子细胞中会出现大量畸形的染色体,表现出范可尼贫血的各种症状。
同时,遗传物质没法正常修复,还会让人体细胞更容易发生致癌突变。因此,范可尼贫血患者比一般人更容易发生多种癌症。
如果孩子只有一份突变的BRCA2基因,那么就不会患上范可尼贫血,但成年后会比一般人更容易得乳腺癌、卵巢癌或胰腺癌。
其它21个和范可尼贫血有关的基因基本上也是类似的情况。简而言之,超过98%的范可尼贫血患者都在同一个基因上累积了两份突变,导致遗传物质的修复出现问题。
像王萍的孩子们接二连三出现范可尼贫血,就说明她和丈夫很有可能各携带一份同样的突变基因,正好都遗传给了孩子。
另外还有一种情况,是孩子从父母中的一方遗传到了突变基因,同时,另一份正常的基因在发育过程中也出现了突变,导致孩子拥有了两份突变基因,患上范可尼贫血。
当然,除非是近亲结婚,否则父母双亲同时携带同一个基因突变、并遗传给孩子的几率并不高;而孩子在遗传中获得特定基因上新突变的概率更小。因此,范可尼贫血非常罕见。
怎么知道自己得的
是不是范可尼贫血?
范可尼贫血患者的特征是,身体里的各种血细胞都比健康人少,因此会发生各种症状:白细胞少,导致他们容易感染疾病;红细胞少,于是他们往往脸色苍白,容易疲惫;血小板少,则使他们经常出血不止,身上常有淤青。
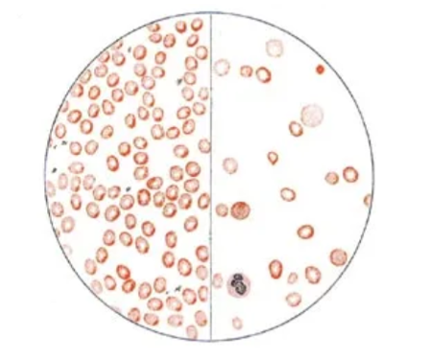
图3 图中左半边是健康人的血液,其中红细胞的数量、大小、形态都正常;
右半边为范可尼贫血患儿的血液,不仅红细胞偏大、数量极少,而且形态不规则。
在大多数范可尼贫血患者中,这些症状会在3-14岁间开始出现,患者平均寿命为20-30岁。不过,不同患者的病情严重程度不一:尽管有约20%的患者活不到成年,但也有不少人活到了30、40甚至50多岁。
超过60%的患者会表现出至少一种以下症状:
- 骨骼(尤其是臀部、脊柱和肋骨)出现问题导致脊柱侧凸,表现为脊柱弯曲;
- 皮肤颜色异常,或出现色素沉着或白癜风;
- 耳部发育异常导致耳聋,眼睛或眼睑出现问题,头围小,个子小;
- 心脏、肺、消化道、肾脏和生殖器发育异常;
- 手臂和手部发育出现问题,例如出现多指或手指形状奇特,或者前臂的骨头很小或干脆没有。
典型范可尼贫血患者的手部X光片。
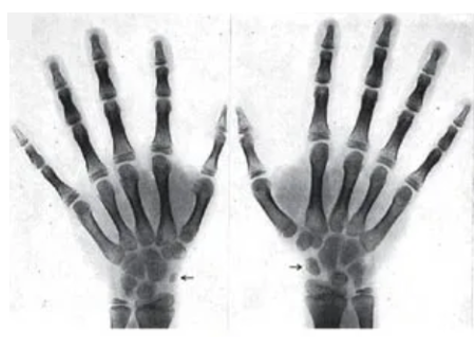
图4 箭头指出的是手腕,那里骨骼发育不全,导致拇指偏短。
其他可能出现的症状还有:
- 出生体重低
- 智力发育异常
- 学习障碍
这些症状可以通过血常规检查、手部X光、CT、核磁共振(MRI)、听力测试、发育测试、肾脏超声检查、骨髓穿刺和活检等方式来确认。
同时,疑似患者还需要进行细胞遗传学检查:这种检查会用一些化学药剂,促使样本细胞中的遗传物质出错,看看这些细胞能否像正常人细胞那样,及时修复这些错误——这是范可尼贫血确诊过程中重要的一环。
得了范可尼贫血该怎么办?
确诊范可尼贫血后,条件允许的话,患者和家族成员首先可以进行遗传咨询。
遗传咨询包括两方面:一方面对患者进行基因检测,确定具体的突变基因,这对范可尼贫血的分型及后续治疗非常重要。另一方面,也要对家族成员进行基因排查。
由于范可尼贫血需要父母双方的基因都有突变才会发病,因此患者的亲属虽然没有患上范可尼贫血,但也很可能携带基因突变,而这些突变都与遗传物质修复相关,因此他们得癌症的可能性还是会比一般人更高,要注意及早筛查。
目前,范可尼贫血还没有特效药。要想长期控制贫血症状,唯一的办法是进行造血干细胞移植,也就是俗称的骨髓移植。即使移植后,患者全身除了血液以外,其他器官的基因还是存在缺陷,因此仍然比普通人更容易得癌症。
对于轻中度的患者,目前主要是通过短期治疗来控制症状,包括:
- 定期就诊检查,并且每年进行骨髓穿刺;
- 筛查(各种类型的)肿瘤和癌症;
- 用抗生素治疗反复感染;
- 有些患者可能会需要输血。
对于病情严重的患者,如果不进行造血干细胞移植,那么为了提升长期的生活质量,延长生存期,可以考虑:
- 使用一些生长因子,如促红细胞生成素、G-CSF和GM-CSF(后两种属于俗称的“升白针”),以便暂时提升血细胞的数量;
- 使用雄性激素进行治疗,雄性激素可以帮助身体制造更多的红细胞和血小板,但对于提升白细胞数目效果不明显;
对于范可尼贫血造成的一些先天生长畸形,可以通过手术治疗。
科学家们也在研究更有效的治疗方法。2012年,美国科学家启动了一项治疗范可尼贫血的基因疗法临床试验,针对的是一个叫做FANCA的突变。
如果这项疗法获得成功,就可以修正突变基因,从根本上治愈疾病。这项研究目前还在进行中。
除了治疗以外,范可尼贫血的患者还需要尽量避免非必要的放射性检测(如X光、CT等)或治疗。
这是因为放射会诱导我们的遗传物质出现错误,而范可尼贫血患者由于基因突变,修复错误的能力远远不足,所以他们的身体对这些放射线非常敏感,容易因为过量的放射而诱发癌症等疾病。
结语
尽管范可尼贫血目前还没有特效的药物或疗法,但它并不意味着绝望。毕竟:
1927年以前,没有人知道这是什么怪病。
1965年以前,科学家们也不清楚究竟是什么导致这种怪病。
1992年以前,没有人知道致病基因是什么。
而现在,2020年,我们已知的范可尼贫血致病基因就多达22个。
而且,随着产前检测技术的发展,即使携带范可尼贫血相关的突变,也可以通过产前检测(羊膜穿刺)或试管婴儿技术胚胎植入前的遗传学诊断,来辅助生育健康的孩子。
随着人类对生命的深入理解和生物技术的革新,我们对致病基因的了解会越来越深,离真正的治愈性疗法也会越来越近,又有什么理由不抱希望呢?

图5
参考文献:
1.Lobitz S, Velleuer E. Guido Fanconi (1892-1979): a jack of all trades. Nar Rev Cancer 2006; 6: 893-8.(本文配图来源)
2.Hays LE, Meyer S, van der Vrugt HJ. A molecular, genetic, and diagnostic spotlight on Fanconi anemia. Anemia Vol 2012, doi:10.1155/2012/650730
3.Guido Fanconi. https://en.wikipedia.org/wiki/Guido_Fanconi
4.Fanconi Anemia: Guidelines for Diagnosis and Management. 4th ed. 2014. Fanconi Anemia Research Fund, Inc. https://www.fanconi.org/images/uploads/other/FARF_Guidelines_Book_interior_lo-res.pdf
5.https://clinicaltrials.gov/ct2/show/NCT01331018
作者 | 徐佳琳
编辑 | 严青
排版 | 狗儿
校对 | 狗儿
相关文章
从不治之症到九成治愈,二百年间人类付出了怎样的努力?
有一种肿瘤专门侵袭宝宝的眼睛,甚至会让孩子失去他们的眼睛,乃至生命。
成都市妇女儿童中心医院儿童血液肿瘤科就诊指南
今天为大家介绍成都市妇女儿童中心医院儿童血液肿瘤科的就诊指南。
【科普基地】好消息!小儿肿瘤科普教育基地新增4个科室,覆盖15个城市23个科室
今年,小儿肿瘤科普教育基地又添4个新成员!
小白专场来了!苏州儿童医院血液科肖佩芳老师等你来提问!
小白专场来了!苏州儿童医院血液科肖佩芳老师等你来提问!| 向日葵问答
四川大学华西第二医院儿童血液肿瘤科就诊指南
向日葵儿童推出了“医院就医指南”项目,今天为大家带来四川大学华西第二医院儿童血液肿瘤科的就诊指南。







